
Development
 Marching cubes 33 C and C++ libraries (Aug. 2019). Further information here.
Marching cubes 33 C and C++ libraries (Aug. 2019). Further information here.
If you use the MC33-UC program, please cite this paper: Vega, D., Abache, J., Coll, D., A Fast and Memory-Saving Marching Cubes 33 Implementation with the correct interior test, Journal of Computer Graphics Techniques (JCGT), vol. 8, no. 3, 1-18, 2019
AIM-UC v1.6.5 (Apr. 2019): For Win32 and Win64 operating systems.
MC33-UC v1.5.3 (Apr. 2019): For Windows 64 bits operating system.
AIM-UC v1.6.4 (May. 2018): For Win32 and Win64 operating systems. Fixed small bugs. This version was also compiled for macOS Version 10.12.6 (Sierra) by Sebastian Potthoff.
 |
MC33-UC v1.5.2 (May. 2018): For Windows 64 bits operating system.
 |
AIM-UC v1.6.2 (Apr. 2017): For Win32 and Win64 operating systems.
Some bugs were fixed. Option to save surfaces were added (the binary *.sup file can be read with MC33-UC)
 |
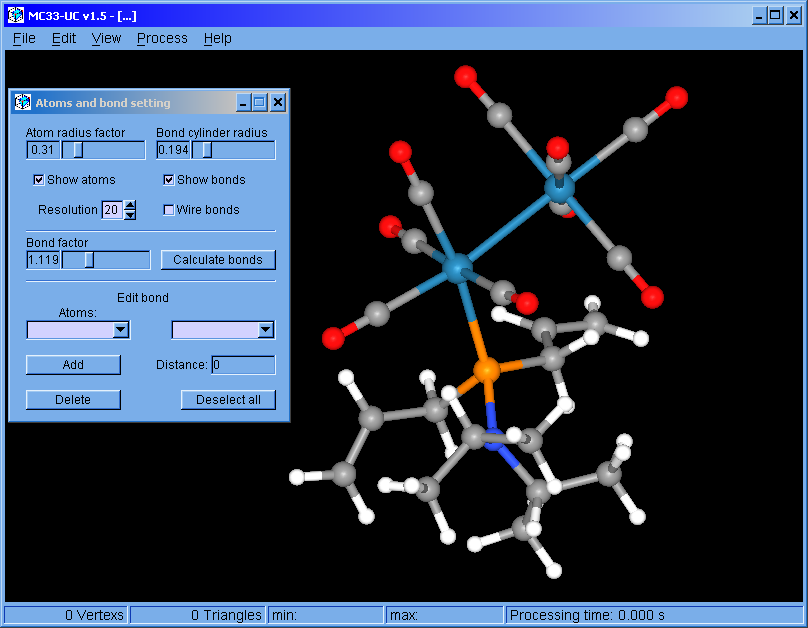
MC33-UC v1.5 (Feb. 2017): For Windows 64 bits operating system.
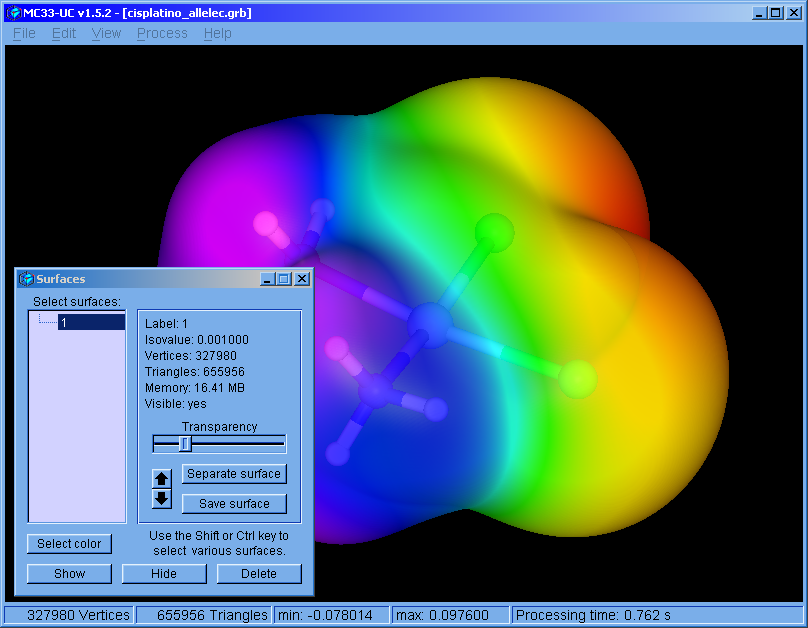
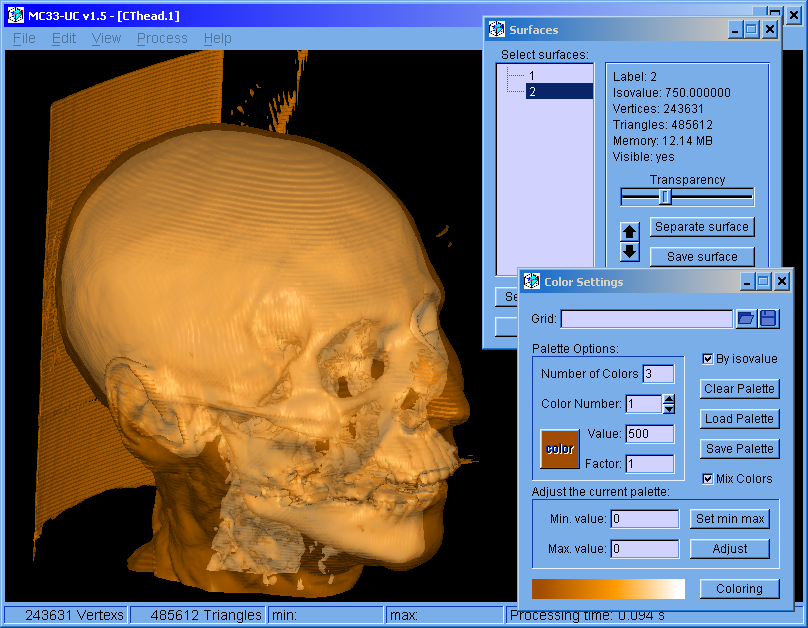
The program was restructured. This version can handle multiple surfaces. The surfaces can be colored independently. Molecular bonds can be modified.
 |
 |
AIM-UC v1.6 (Oct. 2016): For Win32 and Win64 operating systems.
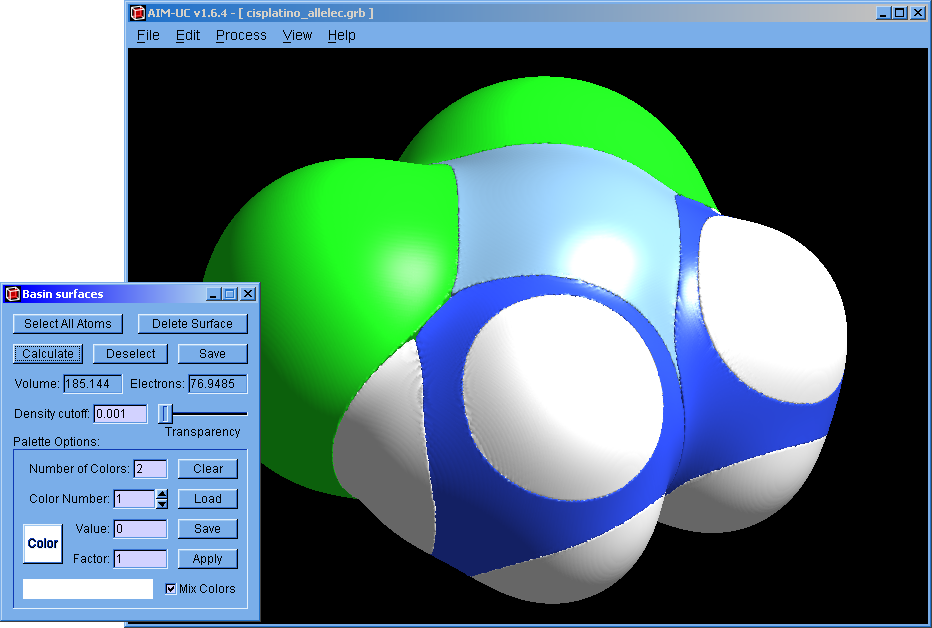
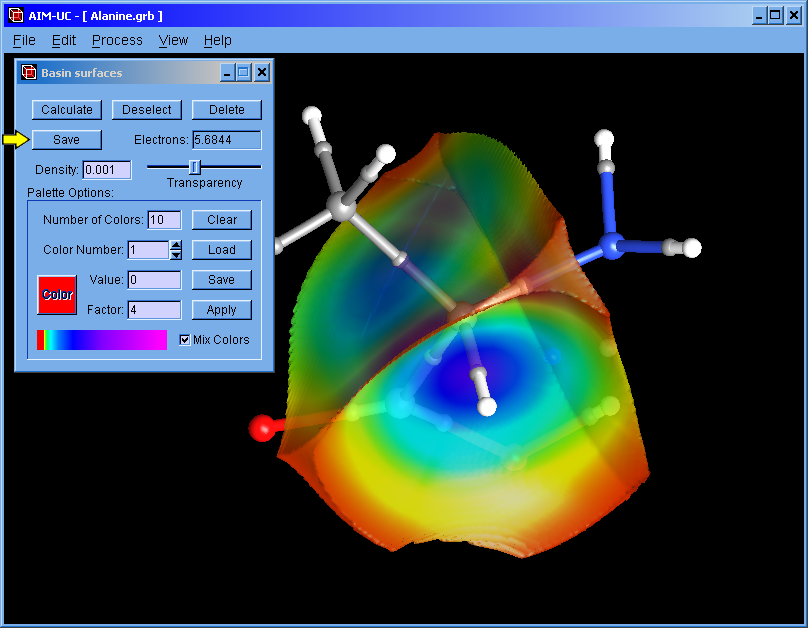
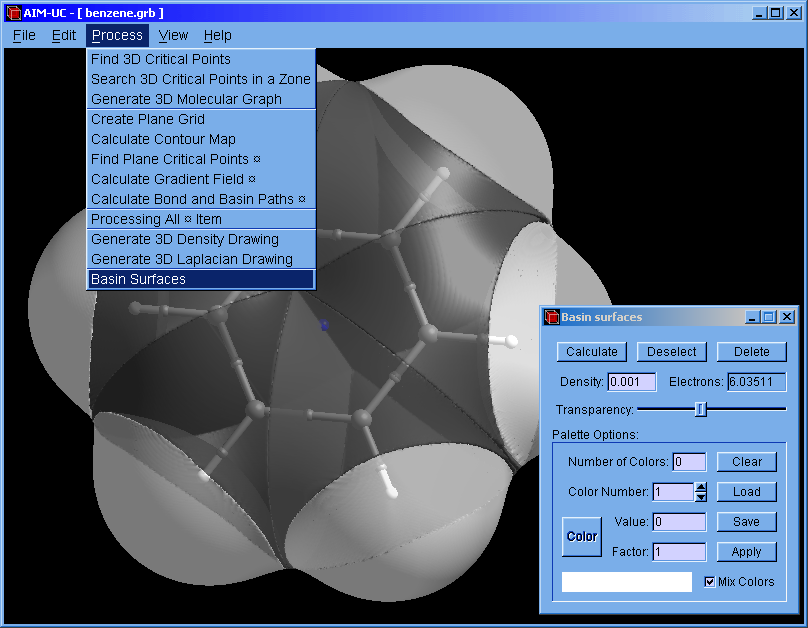
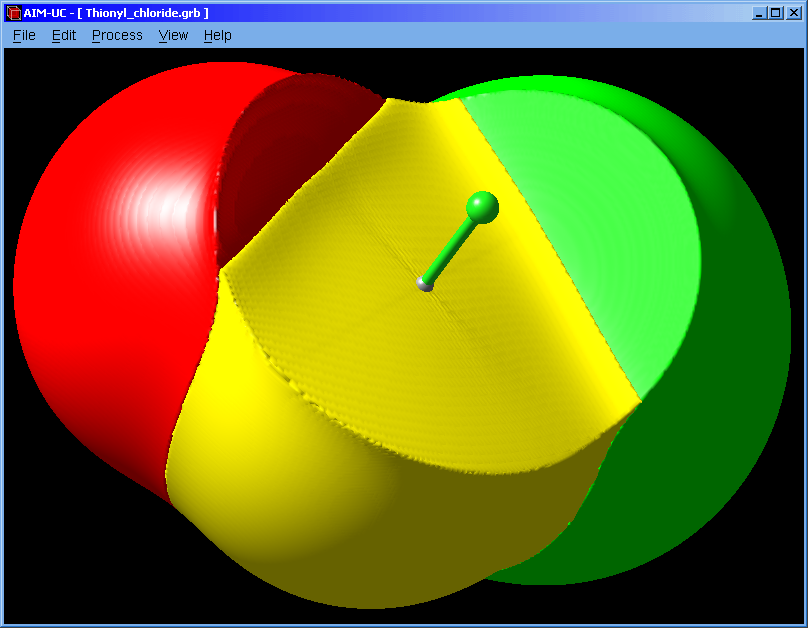
An approximation to QTAIM basins was added to this version. Works for grid data files (grd from DMol, CUBE from Gaussian or CHGCAR from VASP). Use the wfn to binary program to calculate a grid from an AIMPAC file (*.wfn).
Usage:
1. Read the grid data file: Press menu "File", "Open File", or drag and drop the file on the AIM-UC graphics window.
2. Open the "Basin surfaces" window: Press menu "Process" and "Basin Surfaces".
3. With the mouse select an atom (or several atoms) and press the "Calculate" button.
When a group of atom is selected, the calculated basin does not contains the interatomic surface between adjacent atoms.
The basin can also be selected with the mouse. The selected basin can be colored according to the electron density by using a palette (which can be saved and loaded from a file). When the palette is empty, basins can be colored with different colors by pressing the "Apply" button.
 |
 |
AIM-UC v1.5 (Nov. 2015): For Win32 and Win64 operating systems.
Integration of electron density.
The integration is done using the method proposed by Min Yu and Dallas R. Trinkle, but the algorithm was modified: The routine that calculates weights is recursive, marking the grid points when needed, this permits to avoid the process of ordering the grid points by their density value (less memory usage). Also the marking is done for a single atom, so only one weight value for each grid point is necessary. The time to integrate a single atom is comparable to integrate all atoms, but the additional memory needed is much less than the required by the original algorithm.
To perform the integration you must have a grid (grd from DMol, CUBE from Gaussian or CHGCAR from VASP). For AIMPAC files (*.wfn), you can create a grid using MC33-UC or wfn to binary.
Usage:
1. Read the grid data file: Press menu "File", "Open File", or drag and drop the file on the AIM-UC graphics window.
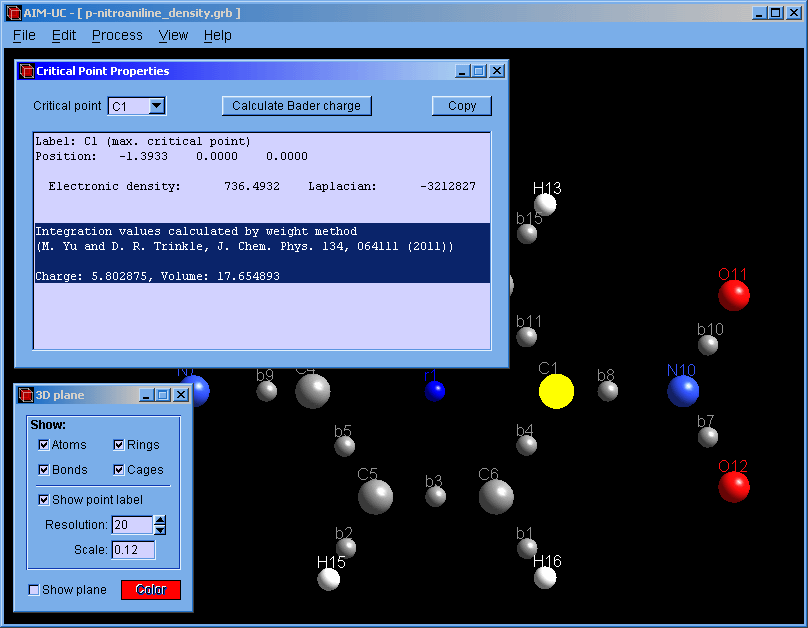
2. The critical points must be calculated (only the maxima are necessary for integration). Press menu "Process" and "Find 3D Critical Points".
3. Open the "Critical Points Properties" window: Press menu "View" and "3D critical point properties".
4. With the mouse select the atom and press the "Calculate Bader charge" button.
 |
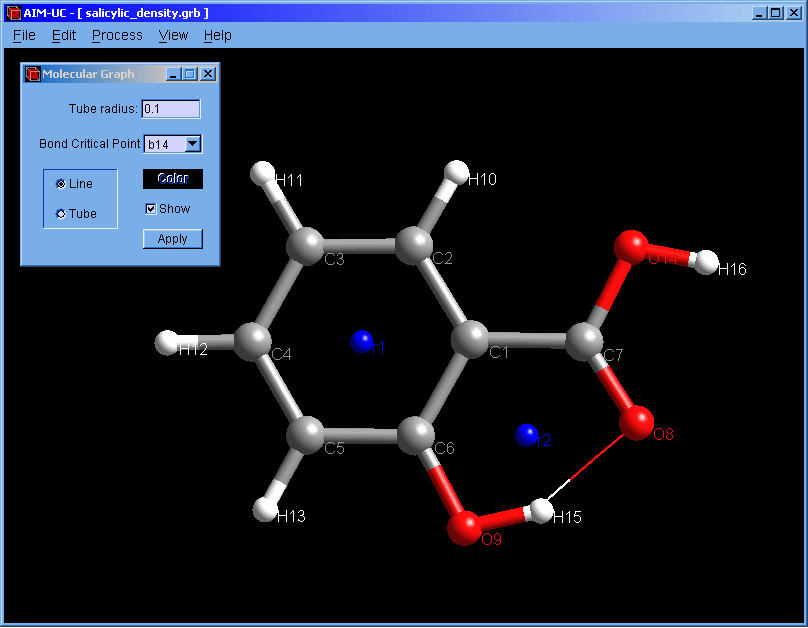
New version of AIM-UC (v1.4): This version include 3D molecular graph calculation (Process, Generate 3D Molecular Graph) and properties of critical points (View, 3D critical point properties). Graphics have been improved. Atoms and critical points can be selected by clicking with the mouse in the graphical window. For now, the program is available for Windows operating systems: Win32 and Win64. Soon we will add a brief description of these features in the manual and compile AIM-UC for some Linux operating systems.
 |
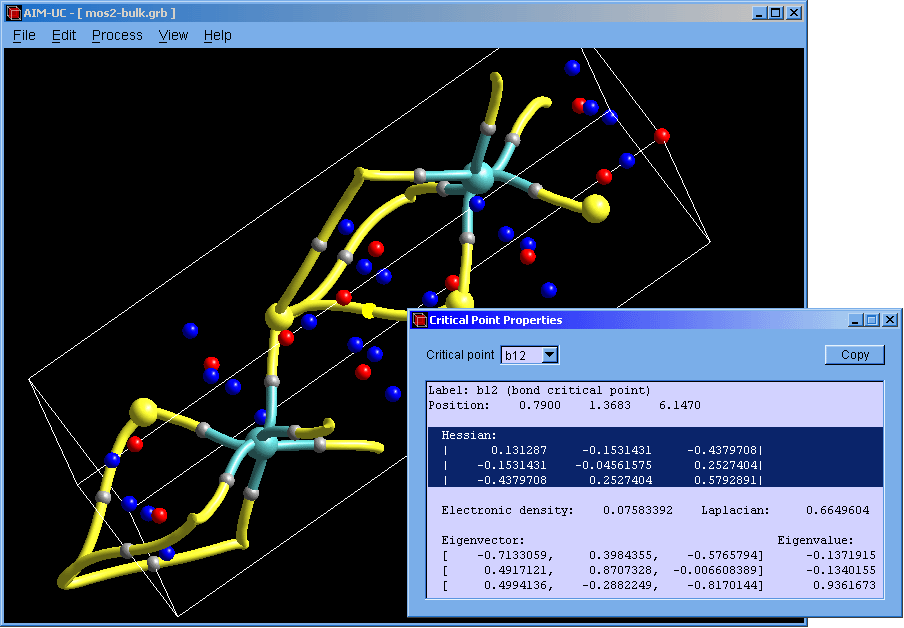 |
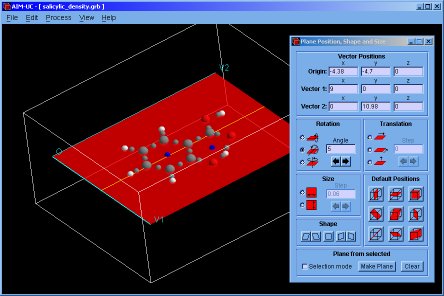
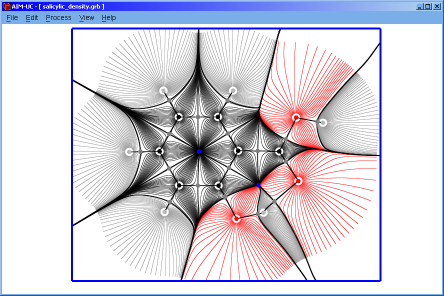
♦ AIM-UC:
Program that generates some graphs related to Bader's AIM theory (theory of atoms in molecules) in a plane in 3D space. The input consists in AIMPAC wavefunction files or numerical grid data of electronic charge density. It can work with huge size grids. It was implemented in C programming language using fltk and OpenGl as external libraries.
A manual of the program wrote in html language can be downloaded in english or spanish.
Now available for 64 bits Linux: Ubuntu 12.10, Xubuntu 12.10 and Centos 6.3.
If you want to cite the program, please cite this paper: "AIM-UC: An application for QTAIM analysis"
 |
 |
♦ Charge integration by weight method:
A kind of Bader integration. When several grid points belong to two or more atomic basins. This method was developed by Min Yu and Dallas R. Trinkle.
Disadvantages: This method uses too much memory, about six times the occupied by the original grid. There is no clear separation between the atomic basins.
Advantages: It is the fastest method available. The result of the integration does not depend on user-defined parameters.
Usage: Put the exe file in a folder. Drag and drop the grid file (in format cube, chgcar, grd or binary) over the executable in the window explorer. A text file with the results will be created in the folder that contains the grid.
♦ wfn to binary:
This application create a grid in binary format (an internal format, based on the grd format of dmol3) starting from a wfn file. This grid is necessary to integrate the Bader charges.
Usage: Put the exe file in a folder. Drag and drop the wfn file over the executable. A grid will be created in the folder that contains the wfn file. An ini file will be created in the executable folder. In this last file you can modify the grid spacing and the distance from the edges of the grid to the outer atoms.
♦ C Library for Topological Study of the Electronic Charge Density (Updated on: Friday, April 19, 2013): The topological study of the electronic charge density is useful to obtain information about the kinds of bonds (ionic or covalent) and the atomic charges on a molecule or crystal, according to the theory of atoms in molecules (AIM). For this study, it is necessary to calculate the electronic density value in every space point, and all electronic density derivatives up to the second order.
The library is based on a multidimensional Lagrange interpolation in a regular grid. It was implemented for 3 dimensions. By differentiating the resulting polynomial, it is possible to obtain, for each space point, the gradient vector, the Hessian matrix and the Laplacian. Functions such as the Newton-Raphson method (to find the critical points, those points where the gradient is null) and the Cash-Karp Runge-Kutta method (to make the gradient paths) were programmed.
Since in some crystals the unit cell has angles different from 90°, the described library includes linear transformations to correct the gradient and Hessian when the grid is distorted (inclined).
Functions were also developed to handle grid data files (grd from DMol® program, CUBE from Gaussian® program and CHGCAR from VASP® program). Each one of these files contains the data of a molecular or crystal electronic property (such as charge density, spin density, electrostatic potential, and others) in a three-dimensional grid. The library can be adapted to make topological studies in any regular three-dimensional grid by modifying the code of these functions.
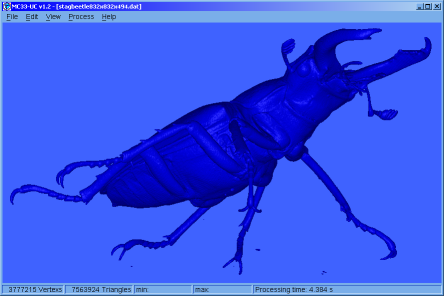
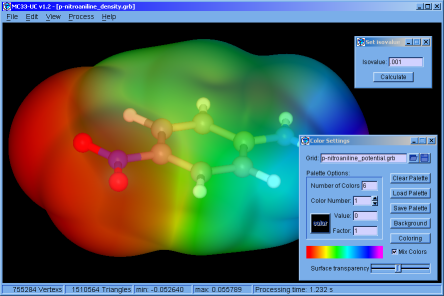
♦ MC33-UC:
Simple application that allows calculating, colouring and displaying isosurfaces, from files that contain 3D grids. The application has tools for managing electron density grids, and also generates grids from the * .wfn files.
Was made using FLTK and our Marching Cubes 33 library. (The manual is not yet available and the version only works on Win64 operating systems)
 |
 |
♦ MARCHING CUBES: Correct and Efficient Implementation: A Marching Cubes 33 (MC33) method based on algorithm that calculates isosurfaces was implemented in C programming language. It is fast, it does requires no great amount of memory and the data type of the grid can be modified.
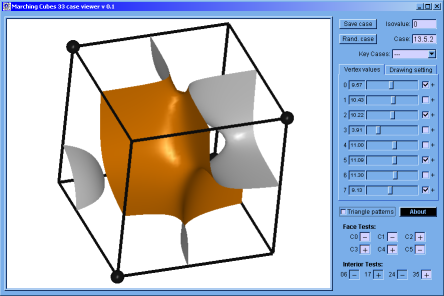
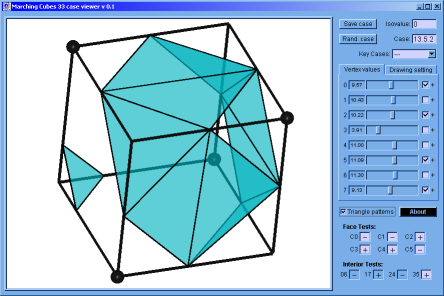
♦ Marching Cubes Case Viewer: It's a program implemented in C programming language; to verify that the Marching Cubes version built by us works correctly. Here, is possible to appreciate the topology of any case and to get the result of the interior and faces test (if this apply).
 |
 |
Address and contact:
Universidad de Carabobo, Av. Salvador Allende, Edif. FACYT - Química.
Bárbula - Edo.Carabobo.
Venezuela.
contact e-mail: Prof. David Vega 